
- Ruhr-Universität Bochum

Atomic cluster expansion for PT-RH catalysts
Noble metals platinum (Pt) and rhodium (Rh) have been extensively employed as heterogeneous catalysts in various applications, such as vehicle exhaust catalysts and fuel cells, due to their remarkable activity and stability. These catalysts are often used in the form of nanoparticles, which maximize the active surface area for chemical reactions. Notably, the internal microstructure of these nanoparticles can be tailored to enhance their functional properties and structural stability. One popular approach involves creating core-shell nanoparticles, where a core of one metal (e.g., Rh) is enveloped by a shell of another metal (e.g., Pt). This core-shell design can boost catalytic activity and stability.
However, maintaining the stability of non-equilibrium nanoparticles under operational conditions can be a challenge. Atomic-scale simulations can provide deeper insights into the thermodynamic and kinetic phenomena governing the structure of these nanoparticles. These simulations can provide information about the energetic aspects of specific structural features as well as the dynamical evolution of the entire system. However, to perform accurate simulations, reliable interatomic interaction models are essential.
The atomic cluster expansion (ACE), developed at ICAMS by Prof. Drautz and his colleagues [1-3], presents a state-of-the-art approach for description of interatomic interactions. ACE combines machine learning methods and physically based models of interatomic interactions, offering superior accuracy and computational efficiency. One of the key features of ACE is a complete and hierarchical set of basis functions that span the space of local atomic environments. This enables to expand an atomic property, such as the energy of atom, with outstanding accuracy.
Recently, we have developed an ACE parametrization for the Pt-Rh system using a semi-automatic workflow [4]. This workflow involves generating a training dataset based on density functional theory (DFT) calculations, fitting the model, and carrying a series of fundamental validation tests. The workflow also employs active learning (AL) algorithms to improve the model’s accuracy. The parameterization of ACE models was based on about 30 000 reference DFT calculations. The considered atomic structures span a wide portion of the configuration space for both elemental metals and their binary compounds. After initial training, AL was employed to ensure a reliable description of Pt-Rh surfaces as these are crucial for simulations of core-shell nanoclusters.
The validation of the parameterized potential included various fundamental properties of the elemental metals as well as the phase stability and other thermodynamic properties of various Pt-Rh phases.
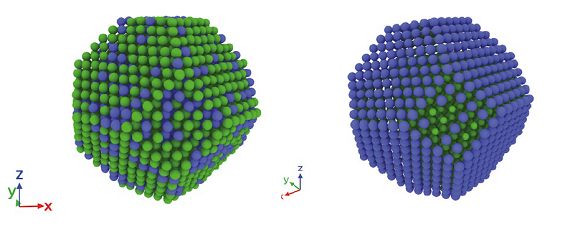
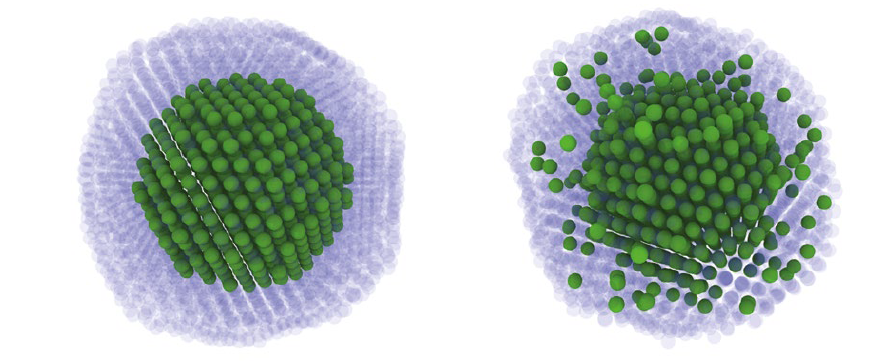
Finally, the ACE parametrization was applied in a series of atomistic simulations to investigate structural stability of various Pt-Rh nanoclusters. The primary focus of the simulations was to examine the role of surface segregation that may play a role in stabilization of the core-shell geometry. Largescale molecular dynamics (MD) and Monte Carlo (MC) simulations were used to explore the thermodynamic stability and the dynamical evolution of realistic clusters at various temperatures and chemical compositions. We observed a strong tendency of the Pt atoms to segregate at cluster surfaces and to form a single monolayer coverage of the whole cluster (see Figure 1). While the formation of a Pt surface layer can be related to low surface energies of Pt, experimental observations of the core-shell particles show a thicker coverage of 3-5 layers of Pt at the surface. By carrying out additional simulations at elevated temperatures (see Figure 2), we showed that the core-shell cluster morphologies consisting of a Rh core with a thicker Pt shell observed in experiments are not thermodynamically favorable but rather kinetically stabilized. This corroborates with the fact that the experimental synthesis procedure creates the core first followed by the coating of the surface element. However, the ACE predictions may be also affected by the limited accuracy of the DFT calculations for Pt surface energies. It is therefore necessary to reexamine the defect energetics in Pt using advanced DFT functionals in order to fully resolve this issue.
REFERENCES:
[1] Drautz R., Physical Review B (2019), 99, 014104.
[2] Bochkarev A., et al., Physical Review Materials (2022), 6, 013804.
[3] Lysogorskiy Y., et al., npj Computational Materials (2021), 7, 97.
[4] Liang Y., et al., Journal of Materials Research (2023).
Dr. Yanyan Liang