
- Ruhr-Universität Bochum
Atomic Cluster Expansion for Large-scale Atomistic Simulations with Quantum Accuracy
Electronic structure calculations have become a valuable tool for materials research and nowadays take a prominent place in many research projects. However, their high computational cost still presents a major limitation and simulations for only a few hundred atoms and picosecond time scales require access to supercomputers. This limits modeling of many important phenomena in materials science, chemistry, physics, and biology, for instance, predictions of phase diagrams and phase transformations, behavior of extended defects, diffusion, material degradation, etc. To be able to simulate these phenomena one has to invent effective interaction potentials that mimic closely the energies and forces from electronic structure calculations, but at a much lower computational cost. By adopting methods from machine learning (ML) and data science, tremendous progress could be achieved in recent years. To construct a ML interatomic potential, one starts by carrying out large numbers of automated high-throughput density functional calculations (DFT), typically for tens of thousands of different structures. The DFT database is then used to train the ML potential by varying its parameters to match the reference data as closely as possible. After careful validation, the ML interatomic potential is ready for large-scale simulations with millions of atoms and simulation times of nanoseconds.
We have recently developed the Atomic Cluster Expansion (ACE) and demonstrated that it shifts previously established limits of ML interatomic potentials towards more accurate and numerically more efficient simulations. The method is general and suitable for most elements and compounds across the periodic table. Efficient implementations for CPU and GPU hardware are available from ICAMS and have already been incorporated in prominent simulations codes, such as LAMMPS.
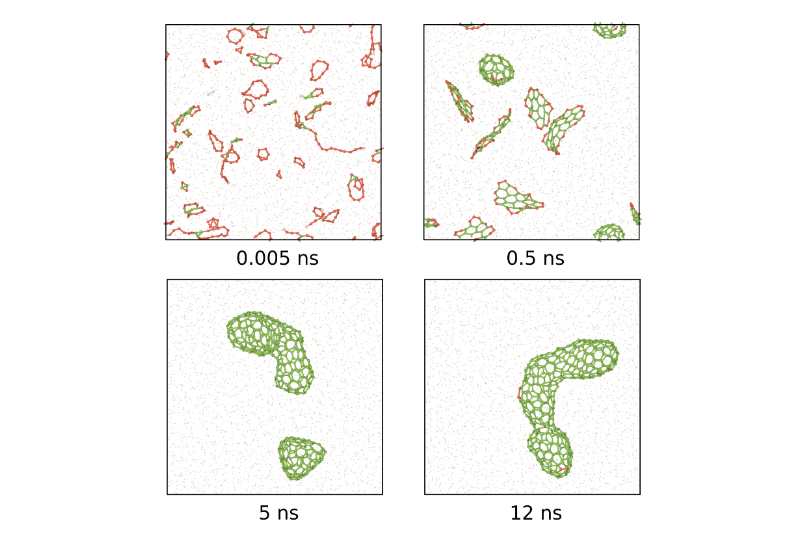
The figure above illustrates a long time-scale simulation of nucleation and growth of carbon fullerenes. Initially, carbon atoms are distributed randomly in an argon atmosphere under high pressure and at high temperature. Forces between the atoms are predicted by ACE and the acceleration is integrated into a trajectory for each atom. From left to right, one first observes the nucleation of small carbon clusters that gradually coalesce and form fullerene molecules. At the end of the simulations only a single large fullerene molecule remains. The same ACE potential that predicts fullerene formation and growth can be used for diverse simulations of amorphous carbon, crack propagation in diamond, or defects in graphene, thereby providing a simulation tool that is able to cross the boundaries between materials science, chemistry, physics and biology.